代谢数据库拓展
2024-09-17 · 25 min read
代谢相关数据库拓展
目前已完成了初步注释,使用eggnog数据库,得到3964条基因(带有EC号)。
后续工作分为两个步骤:1. 对初步结果进行手动验证,有的基因可能与代谢关系不那么直接,可适当删除。2. 查阅文献,拓展数据库进行注释,与初步结果进行比较分析,最终得到全面而严格的代谢相关基因数据集。
# 常见数据库
1. HMDB:人体代谢组数据库(HMDB)是一个免费的电子数据库,包含关于人体小分子代谢产物的详细信息,其应用领域包括代谢组学、临床化学、生物标志物的发现。该数据库包含114,026个代谢物记录,包括水溶性和脂溶性代谢物,以及被认为是丰富(> 1 uM)或相对稀少(< 1 nM)的代谢物。此外,还有5702个蛋白质序列与这些代谢产物相关。
http://www.hmdb.ca/
2. KEGG:东京基因及基因组百科全书,全书收录了一只各种生物的所有代谢物的代谢途径。支持对代谢网络的搜寻及代谢途径的映射。与代谢组学相关性大的几个模块包括:KEGG PATHWAY,KEGG DISEASA,KEGG COMPOUND,KEGG REACTION。
https://www.genome.jp/kegg/ligand.html
3. Reactome:REACTOME 是一个开源、开放访问、手动策划和同行评审的途径数据库。其中包含信号传导和代谢分子及其组织成生物途径和过程的关系。Reactome 数据模型的核心单元是反应。参与反应的实体(核酸、蛋白质、复合物、疫苗、抗癌疗法和小分子)形成生物相互作用网络,并分为通路。Reactome 中的生物通路示例包括经典中间代谢、信号传导、转录调控、细胞凋亡和疾病。(与KEGG互补)
分析参考:https://blog.csdn.net/weixin_43839173/article/details/125318973
https://reactome.org/
# 0911更新:放弃该数据库,本地注释没有特别好的方式。
# 其它数据库,参考系统生物学paper
1. BiGG Models:是由美国University of California, San Diego 创立的基于代谢组学的系统生物学整合数据库。 该数据库的最大特点是含有各类模式生物的代谢谱图模型。用户可以直观的调取各种生物的整体代谢通路,也可以查看某个具体的生化反应。同时也可以进行代谢产物搜索。该数据库目前含有2766个代谢产物和3311条代谢生化反应。
http://bigg.ucsd.edu/
ref:https://doi.org/10.1371/journal. pcbi.1009870
2. BRENDA:是一种专门用于酶和代谢酶的功能信息的数据库。它是一种在线资源,提供了大量的酶的相关信息,如酶的命名、分类、反应类型、催化物质、底物和产物等。Brenda数据库还包含了酶的催化机制、反应动力学、酶的结构和序列信息等。
https://www.brenda-enzymes.org/
doi:10.1038/nprot.2009.203
# 240911更新,没必要一味地扩大数据库,追求方法学上的完美,因此这两个数据库被放弃,我们要尽早得到基因数据集开展实验。
初步挑选了6个数据库,探索其本地注释的可行性。
0911更新:确定eggnog,kegg和hmdb三个数据库用作分析。
1. HMDB(官网蛋白文件下载界面:https://hmdb.ca/downloads)
# sed -i 's/旧词/新词/g' 文件名 (fasta格式不规范,没有以>开头)
sed -i 's/HMDBP/>HMDBP/g' hmdb.fasta
# 发现下载的蛋白文件只有5629条序列,而官网上却显示8299条,已联系团队咨询此问题,暂时用下载文件分析。
grep "HMDB" hmdb.fasta |wc -l
5629
# cd-hit检测该数据库的冗余度,发现95%相似度下,只有几百条冗余,不愧是人类代谢数据库!
makeblastdb -in hmdb.fasta -dbtype prot -title hm -parse_seqids -out ./hm
nohup blastp -query /data/xb/1_pig_genome/GCF_000003025.6_Sscrofa11.1_protein.faa -db ~/0_rawdata/database/hmdb/hm -max_target_seqs 1 -outfmt 6 -evalue 1e-5 -num_threads 8 > pig.hm.tab &
cat pig.hm.tab|cut -f1 > id.list
sort -n id.list|uniq > rmdup.list
#删除没意义的行 “Warning...”
sed '/Warning/d' rmdup.list > delete.rmdup.listq #得到的蛋白序列多达38706条,而eggnog仅有12539条。
可能要上双向最佳比对,否则得到的基因数太多了(测试发现单向比对最后得到gene序列12458条,而eggnog仅有3694条,更符合文献报道)
双向最佳比对,用猪蛋白建库
mkdir pig && cd pig
makeblastdb -in ../GCF_000003025.6_Sscrofa11.1_protein.faa -dbtype prot -title pig -parse_seqids -out ./pig
nohup blastp -query ~/0_rawdata/database/hmdb/hmdb.fasta -db /data/xb/1_pig_genome/pig/pig -max_target_seqs 1 -outfmt 6 -evalue 1e-5 -num_threads 8 > hmdb.pig.tab &
sed -i '/Warning/d' hmdb.pig.tab
# 双向最佳比对
import pandas as pd
# 读取正向BLAST和反向BLAST的比对结果,指定文件路径
forward_blast_path = '/home/xb/1_results/240906_pig_genome_annot/hmdb/pig.hm.tab'
reverse_blast_path = '/home/xb/1_results/240906_pig_genome_annot/hmdb/rbh/hmdb.pig.tab'
# 读取BLAST结果文件,假设为outfmt 6格式
blast_forward = pd.read_csv(forward_blast_path, sep='\t', header=None)
blast_reverse = pd.read_csv(reverse_blast_path, sep='\t', header=None)
# 设置列名
columns = ['qseqid', 'sseqid', 'pident', 'length', 'mismatch', 'gapopen', 'qstart', 'qend', 'sstart', 'send', 'evalue', 'bitscore']
blast_forward.columns = columns
blast_reverse.columns = columns
# 筛选正向和反向的 query 和 subject 组合,并确保唯一性
best_hits_forward = blast_forward[['qseqid', 'sseqid']].drop_duplicates()
best_hits_reverse = blast_reverse[['sseqid', 'qseqid']].drop_duplicates()
# 找到双向最佳比对
rbh = pd.merge(best_hits_forward, best_hits_reverse, left_on=['qseqid', 'sseqid'], right_on=['sseqid', 'qseqid'])
# 保存结果为文件,指定保存路径
output_path = '/home/xb/1_results/240906_pig_genome_annot/hmdb/rbh/rbh.tab'
rbh.to_csv(output_path, sep='\t', index=False)
根据蛋白ID从GBFF中提取Gene_id
from Bio import SeqIO
def extract_gene_ids(gbff_file, protein_ids_file, output_file):
# 读取蛋白质ID列表
with open(protein_ids_file, 'r') as f:
protein_ids = [line.strip() for line in f.readlines()]
# 创建一个字典来存储蛋白质ID与基因ID的映射
protein_to_gene = {}
# 解析GBFF文件
for record in SeqIO.parse(gbff_file, "genbank"):
for feature in record.features:
if feature.type == "CDS":
# 提取protein_id
if "protein_id" in feature.qualifiers:
protein_id = feature.qualifiers["protein_id"][0]
if protein_id in protein_ids:
# 提取GeneID
gene_ids = [xref.split(":")[1] for xref in feature.qualifiers.get("db_xref", []) if xref.startswith("GeneID")]
if gene_ids:
protein_to_gene[protein_id] = gene_ids[0] # 只取第一个GeneID
# 输出结果到文件
with open(output_file, 'w') as out_f:
out_f.write("Protein_ID\tGene_ID\n")
for protein_id in protein_ids:
gene_id = protein_to_gene.get(protein_id, "Not found")
out_f.write(f"{protein_id}\t{gene_id}\n")
# 示例使用
gbff_file = "/data/xb/1_pig_genome/GCF_000003025.6_Sscrofa11.1_genomic.gbff" # 替换为你的GBFF文件路径
protein_ids_file = "/home/xb/1_results/240906_pig_genome_annot/hmdb/rbh/rbh.list" # 替换为包含蛋白质ID的txt文件路径
output_file = "/home/xb/1_results/240906_pig_genome_annot/hmdb/extract_from_gbff/gene.id.txt" # 输出结果的文件路径
extract_gene_ids(gbff_file, protein_ids_file, output_file)
# 提取第二列的gene_id并去重
cat gene.id.txt|cut -f2 > id.list
sort -n id.list |uniq > rmdup.gene.list # 最终结果4930条gene
# 0911测试,将猪蛋白fasta进行cd-hit 95%的聚类后,蛋白数量从63575减少到28102条,重新进行双向最佳比对,结果发现得到的蛋白数4929条,和不聚类相差无几,说明聚类不仅可以减少资源浪费,对结果影响也不大。
# 0913测试,将置信值设为1e-10,得到蛋白数为4948条,和1e-5(4953条)差别不大。
import pandas as pd
# 读取两个表格文件
table1 = pd.read_csv('/home/xb/1_results/240906_pig_genome_annot/eggnog/extract_from_gbff/rmdup.list', header=None)
table2 = pd.read_csv('/home/xb/1_results/240906_pig_genome_annot/hmdb/extract_from_gbff/rmdup.gene.list', header=None)
# 重命名列名为 'ID'
table1.columns = ['ID']
table2.columns = ['ID']
# 表1特有内容
unique_table1 = table1[~table1['ID'].isin(table2['ID'])]
# 表2特有内容
unique_table2 = table2[~table2['ID'].isin(table1['ID'])]
# 共有内容
common = table1[table1['ID'].isin(table2['ID'])]
# 确保目标文件夹存在
import os
output_folder = '/home/xb/1_results/240906_pig_genome_annot/diff_compare/gene'
os.makedirs(output_folder, exist_ok=True)
# 将结果保存为文件
unique_table1.to_csv(f'{output_folder}/unique_table1.csv', index=False, header=False)
unique_table2.to_csv(f'{output_folder}/unique_table2.csv', index=False, header=False)
common.to_csv(f'{output_folder}/common.csv', index=False, header=False)
eggnog注释得到3691条gene id,hmdb得到4930条gene id,eggnog特有1130条,hmdb特有2369条,共有2561条。
切割gene id,从ncbi中批量下载gene序列
# 分割脚本,按照500个切割表格,形成多个小表格供后续使用。
import csv
import os
def split_list(lst, n):
"""将列表分割成多个小列表,每个列表最多包含n个元素。"""
for i in range(0, len(lst), n):
yield lst[i:i + n]
def save_split_files(gene_ids, batch_size, output_dir):
"""将基因ID列表分割并保存到多个CSV文件中。"""
if not os.path.exists(output_dir):
os.makedirs(output_dir)
for idx, batch in enumerate(split_list(gene_ids, batch_size), start=1):
file_path = os.path.join(output_dir, f'batch_{idx}.csv')
with open(file_path, 'w', newline='') as csvfile:
writer = csv.writer(csvfile)
for gene_id in batch:
writer.writerow([gene_id])
if __name__ == "__main__":
csv_file_path = "/home/xb/1_results/240906_pig_genome_annot/hmdb/extract_from_gbff/rmdup.gene.list" # 输入CSV文件路径
output_dir = "/home/xb/1_results/240906_pig_genome_annot/hmdb/extract_from_gbff/split_files" # 输出目录
batch_size = 600 # 每个文件的基因ID数量
gene_ids_from_csv = []
with open(csv_file_path, newline='') as csvfile:
reader = csv.reader(csvfile)
for row in reader:
if len(row) > 0:
gene_id = int(row[0]) # 基因ID在CSV文件的第一列
gene_ids_from_csv.append(gene_id)
save_split_files(gene_ids_from_csv, batch_size, output_dir)
# 下载脚本,根据id下载gene序列,有的gene id在性染色体上,对应能查出两条基因序列。
import sys
import csv
import io
import os
from typing import List
from zipfile import ZipFile
from ncbi.datasets.openapi import ApiClient as DatasetsApiClient
from ncbi.datasets.openapi import ApiException as DatasetsApiException
from ncbi.datasets.openapi import GeneApi as DatasetsGeneApi
def download_and_extract_genes(gene_ids: List[int], zipfile_name: str, output_file_name: str):
"""下载基因数据并提取到文件中。"""
with DatasetsApiClient() as api_client:
gene_api = DatasetsGeneApi(api_client)
try:
gene_dataset_download = gene_api.download_gene_package_without_preload_content(
gene_ids,
include_annotation_type=["FASTA_GENE", "FASTA_PROTEIN"], # 选择下载的数据格式
)
with open(zipfile_name, "wb") as f:
f.write(gene_dataset_download.data)
except DatasetsApiException as e:
sys.exit(f"Exception when calling GeneApi: {e}\n")
try:
with ZipFile(zipfile_name) as dataset_zip:
zinfo = dataset_zip.getinfo(os.path.join("ncbi_dataset/data", "protein.faa"))
with io.TextIOWrapper(dataset_zip.open(zinfo), encoding="utf8") as fh:
with open(output_file_name, "a", encoding="utf8") as output_file:
output_file.write(fh.read())
except KeyError as e:
sys.exit(f"File {output_file_name} not found in zipfile: {e}")
def process_csv_files(input_dir: str, output_file_name: str):
"""处理目录中的所有CSV文件并提取基因数据。"""
csv_files = [f for f in os.listdir(input_dir) if f.endswith('.csv')]
if not csv_files:
sys.exit(f"No CSV files found in directory: {input_dir}")
# 确保输出文件存在
open(output_file_name, 'w').close()
for csv_file in csv_files:
csv_file_path = os.path.join(input_dir, csv_file)
with open(csv_file_path, newline='') as csvfile:
reader = csv.reader(csvfile)
gene_ids_from_csv = [int(row[0]) for row in reader if len(row) > 0]
# 使用 CSV 文件名作为唯一标识的一部分
zipfile_name = f"gene_{os.path.splitext(csv_file)[0]}.zip"
download_and_extract_genes(gene_ids_from_csv, zipfile_name, output_file_name)
if __name__ == "__main__":
input_dir = "/home/xb/1_results/240906_pig_genome_annot/hmdb/extract_from_gbff/split_files" # CSV文件所在目录
output_file_name = "combined_protein.faa" # 合并后的输出文件
process_csv_files(input_dir, output_file_name)
解压、重命名及合并fna文件
#!/bin/bash 解压&重命名
# 确保目标文件夹存在
mkdir -p gene_output
# 遍历所有以 gene_batch_ 开头的 zip 文件
for zip_file in gene_batch_*.zip; do
# 提取文件名(去掉扩展名)
base_name=$(basename "$zip_file" .zip)
# 解压指定文件并重命名
unzip -j "$zip_file" ncbi_dataset/data/gene.fna -d ./gene_output/ &&
mv ./gene_output/gene.fna ./gene_output/${base_name}.fna
done
cd gene_output/
cat *.fna > output.fna
seqtk seq -A output.fna | sort -u > output.rmdup.hmdb.fna
grep ">" output.rmdup.hmdb.fna |wc -l # 最后得到4934条gene,结果文件为output.rmdup.hmdb.fna
2. KEGG本地注释
参考:[比较转录组分析(四)—— 组装的 GO 及 KEGG 注释 | Juse's Blog (biojuse.com)](https://biojuse.com/2022/11/28/比较转录组分析(四)—— 组装的注释/)
数据库下载:
mkdir -p /home/xb/0_rawdata/database/kegg/db
cd /home/xb/0_rawdata/database/kegg/db
wget -c ftp://ftp.genome.jp/pub/db/kofam/ko_list.gz
wget -c ftp://ftp.genome.jp/pub/db/kofam/profiles.tar.gz
wget -c ftp://ftp.genome.jp/pub/db/kofam/README
db_Version=`grep "Last update: " README|perl -p -e 's/Last update: //g;s/\//_/g'`
gunzip ko_list.gz
tar xvzf profiles.tar.gz
touch db_Version_${db_Version}
cd ..
wget -c ftp://ftp.genome.jp/pub/tools/kofam_scan/kofam_scan-1.3.0.tar.gz
tar -zxvf kofam_scan-1.3.0.tar.gz
安装依赖
conda create -n kofam -y -c bioconda ruby hmmer parallel
conda activate kofam
cd kofam_scan-1.3.0
cp config-template.yml config.yml
vim config.yml
# 内容如下:
# Path to your KO-HMM database
# A database can be a .hmm file, a .hal file or a directory in which
# .hmm files are. Omit the extension if it is .hal or .hmm file
profile: /home/xb/0_rawdata/database/kegg/db/profiles
# Path to the KO list file
ko_list: /home/xb/0_rawdata/database/kegg/db/ko_list
# Path to an executable file of hmmsearch
# You do not have to set this if it is in your $PATH
hmmsearch: /home/xb/miniconda3/envs/kofam/bin/hmmsearch
# Path to an executable file of GNU parallel
# You do not have to set this if it is in your $PATH
parallel: /home/xb/miniconda3/envs/kofam/bin/parallel
# Number of hmmsearch processes to be run parallelly
cpu: 8
运行命令
mkdir -p /home/xb/1_results/240906_pig_genome_annot/kegg
cd /home/xb/1_results/240906_pig_genome_annot/kegg
ln -s /home/xb/0_rawdata/database/kegg/kofam_scan-1.3.0/exec_annotation kofamscan
./kofamscan -o group_rep.kofam.out --cpu 8 --format mapper -e 1e-5 /data/xb/1_pig_genome/GCF_000003025.6_Sscrofa11.1_protein.faa #测试软件可行性,报错提示Ruby版本太低。
conda install ruby=2.7 #升级至2.7后可运行
nohup ./kofamscan -o group_rep.kofam.out --cpu 8 --format mapper -e 1e-5 /data/xb/1_pig_genome/GCF_000003025.6_Sscrofa11.1_protein.faa &
使用 Kofamscan 得到 KO 注释文件后,可以使用对应物种的 pathway-KO 文件给注释相应的 pathway,编写脚本pathway.py
#!/usr/bin/env python3
import os
import sys
import re
def pathway_map(sp="ko"):
"""
:param sp:The default is'ko', which means downloading 'ko00001.keg'
https://www.genome.jp/kegg-bin/download_htext?htext=ko00001.keg&format=htext&filedir=
:return: K_ko_map
"""
#url = r"https://www.genome.jp/kegg-bin/download_htext?htext=asa00001.keg&format=htext&filedir="
keg_file = sp +"00001.keg"
cmd = r"wget 'http://www.kegg.jp/kegg-bin/download_htext?htext=" +sp + "00001.keg&format=htext&filedir=' -O " + keg_file
if not os.path.exists(keg_file):
try:
res = os.system(cmd)
# 使用system模块执行linux命令时,如果执行的命令没有返回值res的值是256
# 如果执行的命令有返回值且成功执行,返回值是0
except:
print("Failed to run\n" + str(cmd) +"\nplease check the network")
sys.exit()
in_keg = open(keg_file, "r").readlines()
K_ko_map = {}
for line in in_keg:
if line.startswith("A"):
# 'A09100 Metabolism'
level_1 = re.match(r'^A(.+?)\s(.+)\n', line).group(2)
elif line.startswith("B "):
# B 09102 Energy metabolism
level_2 = re.match(r'^B\s*(.+?)\s(.*)\n', line).group(2)
elif line.startswith("C "):
# 'C 00010 Glycolysis / Gluconeogenesis [PATH:asa00010]' or 'C 99980 Enzymes with EC numbers'
pathway_info = re.match(r'^C\s*(\d+?)\s(.*)\n', line)
pathway_id = "ko" + str(pathway_info.group(1))
pathway_desc = str(pathway_info.group(2)).split(" [")[0]
pathway_info_list = [pathway_desc, level_1, level_2]
elif line.startswith("D ") and in_keg[0] == '+D\tGENES\tKO\n':
# 'D ASA_1323 glk; glucokinase\tK00845 glk; glucokinase [EC:2.7.1.2]\n'
K_info = re.match(r'^D\s*.*\t(K\d+?)\s(.*)\n', line)
K_id = K_info.group(1)
K_desc = K_info.group(2)
K_info_list = [K_desc, pathway_id, pathway_desc, level_1, level_2]
if K_id not in K_ko_map.keys():
K_ko_map[K_id] = [K_info_list]
else:
l_tem = K_ko_map[K_id]
if K_info_list not in l_tem:
l_tem.append(K_info_list)
K_ko_map[K_id] = l_tem
elif line.startswith("D ") and in_keg[0] == '+D\tKO\n':
# 'D K00844 HK; hexokinase [EC:2.7.1.1]'
K_info = re.match(r'^D\s*(K\d+?)\s(.*)\n', line)
K_id = K_info.group(1)
K_desc = K_info.group(2)
K_info_list = [K_desc, pathway_id, pathway_desc, level_1, level_2]
if K_id not in K_ko_map.keys():
K_ko_map[K_id] = [K_info_list]
else:
l_tem = K_ko_map[K_id]
if K_info_list not in l_tem:
l_tem.append(K_info_list)
K_ko_map[K_id] = l_tem
return (K_ko_map)
def ko_class_map():
# https://www.genome.jp/kegg-bin/download_htext?htext=br08901.keg&format=htext&filedir= htext
# https://www.genome.jp/kegg-bin/download_htext?htext=br08901.keg&format=json&filedir= josn
# https://www.genome.jp/dbget-bin/get_linkdb?-t+orthology+path:ko00040
cmd = r"wget 'https://www.genome.jp/kegg-bin/download_htext?htext=br08901.keg&format=htext&filedir=' -O br08901.keg"
if not os.path.exists("br08901.keg"):
try:
res = os.system(cmd)
# 使用system模块执行linux命令时,如果执行的命令没有返回值res的值是256
# 如果执行的命令有返回值且成功执行,返回值是0
except:
print("Failed to run\n" + str(cmd) + "\nplease check the network")
sys.exit()
in_keg = open("br08901.keg", "r").readlines()
pathway_map=open("kegg_pathway_map.xls","w+")
ko_class = {}
for line in in_keg:
if line.startswith("A"):
# 'A09100 Metabolism'
level_1 = re.match(r'^A<b>(.*)</b>\n', line).group(1)
elif line.startswith("B "):
# B 09102 Energy metabolism
level_2 = re.match(r'^B\s*(.*)\n', line).group(1)
elif line.startswith("C "):
# 'C 00010 Glycolysis / Gluconeogenesis [PATH:asa00010]' or 'C 99980 Enzymes with EC numbers'
pathway_info = re.match(r'^C\s*(\d+?)\s(.*)\n', line)
pathway_id = "ko" + str(pathway_info.group(1))
pathway_desc = str(pathway_info.group(2)).split(" [")[0]
line_out = pathway_id + "\t" + pathway_desc + "\t" + level_1 + "\t" +level_2 + "\t"
pathway_map.write(line_out + "\n")
pathway_info_list = [pathway_desc, level_1, level_2]
if pathway_id not in ko_class.keys():
ko_class[pathway_id] = pathway_info_list
return(ko_class)
def K_list_Parser(K_list,sp="ko"):
file_name = os.path.split(K_list)[1].rsplit(".",1)[0]
out_kegg_anno = file_name + ".kegg_anno.xls"
not_in_pathway_map = file_name + "K_codes_not_in_pathway_map.list"
out_kegg_pathway = file_name + ".kegg_pathway_stata.xls"
anno_f = open(out_kegg_anno, "w+")
not_in_pathway_f = open(not_in_pathway_map, "w+")
pathway_f = open(out_kegg_pathway, "w+")
anno_f.write("gene_id\tK_id\tK_desc\tpathway_id\tpathway_desc\tlevel_1\tlevel_2\n")
pathway_f.write("Pathway\tGenes annoted in term\tPathway ID\tLevel1\tLevel2\tKOs\tGenes\n")
map_kegg = pathway_map(sp)
ko_class = ko_class_map()
infile_list = open(K_list, "r").readlines()
infile_list = [ term.rstrip("\n").split("\t") for term in infile_list ]
k_num_dict = {}
line_out_tem = ""
for line in infile_list:
gene_id = line[0]
if len(line) == 2:
K_id = line[1]
if K_id in map_kegg.keys():
pathway_info = map_kegg[K_id]
for K_info_list in pathway_info :
# K_desc, pathway_id, pathway_desc, level_1, level_2
string = "\t"
line_out =gene_id + "\t" + K_id + "\t" +string.join(K_info_list) + "\n"
if line_out != line_out_tem:
anno_f.write(line_out)
line_out_tem = line_out
else:
not_in_pathway_f.write(gene_id + "\t" + K_id + "\n")
line_out = gene_id + "\t" * 6 + "\n"
if line_out != line_out_tem:
anno_f.write(line_out)
line_out_tem = line_out
# k_id 2 gene_id list
if K_id not in k_num_dict.keys():
k_num_dict[K_id] = gene_id
else:
k_num_dict[K_id] = k_num_dict[K_id] + ';' + gene_id
else:
anno_f.write(gene_id +"\t"*6 + "\n")
ko_sample_dict = {}
for K_id in list(k_num_dict.keys()):
if K_id not in list(map_kegg.keys()):
continue
ko_num_sample = [ term[1] for term in map_kegg[K_id]]
for ko in ko_num_sample:
if ko not in list(ko_sample_dict.keys()):
ko_sample_dict[ko] = K_id
else:
ko_sample_dict[ko] = ko_sample_dict[ko] + ';' + K_id
for ko in [item for item in list(ko_sample_dict.keys()) if item in list(ko_class.keys()) ] :
pathway = ko_class[ko][0]
level1 = ko_class[ko][1]
level2 = ko_class[ko][2]
k_num_list = ko_sample_dict[ko].split(';')
gene_str = ''
for k_num_sample in k_num_list:
gene_str = gene_str + k_num_dict[k_num_sample] + ";"
num_gene = gene_str.count(';')
# pathway_f.write("Pathway\tGenes annoted in term\tPathway ID\tLevel1\tLevel2\tKOs\tGenes\n")
pathway_f.write(pathway + '\t' + str(num_gene)+ '\t' + ko +'\t' +level1 + '\t'+level2 +'\t'+ko_sample_dict[ko].rstrip(';')+ '\t' + gene_str.rstrip(';')+'\n')
anno_f.close()
pathway_f.close()
if len(sys.argv) < 2: #直接执行本脚本给出帮助信息
print(doc)
sys.exit()
elif len(sys.argv) == 2:
kaas_inflie = sys.argv[1]
K_list_Parser(kaas_inflie)
elif len(sys.argv) == 3:
kaas_inflie = sys.argv[1]
sp = sys.argv[2]
K_list_Parser(kaas_inflie,sp)
else:
print(doc)
sys.exit()
# 特定物种注释,猪ssc
python3 pathway.py group_rep.kofam.out ssc
# 结果说明
br08901.keg kegg pathway分级文件
group_repkofam.kegg_anno.xls 基因K,ko注释(按基因)
group_repkofam.kegg_pathway_stata.xls 基因pathway注释统计(按pathway)
group_repkofamK_codes_not_in_pathway_map.list 没有注释到path的Orthology (K id)
kegg_pathway_map.xls kegg pathway分级表
ko00001.keg 同源簇(KEGG Orthology--KO)信息(参考或特定物种)
# 对group_repkofam.kegg_pathway_stata.xls进一步分析,下载至本地,Level1限制为Metabolism,提取相关的GENE并去重。
编写提取脚本extract.py
import pandas as pd
input_file = '/home/xb/1_results/240906_pig_genome_annot/kegg/metabolism.pro.list'
output_file = '/home/xb/1_results/240906_pig_genome_annot/kegg/metabolism.pro.id'
# 读取只有一列的文件
df = pd.read_csv(input_file, header=None)
# 提取唯一一列(蛋白ID)
protein_ids = df.iloc[:, 0]
# 分割蛋白ID并保存到新文件
with open(output_file, 'w') as outfile:
for protein_id in protein_ids:
# 如果有分号,则按分号分割
ids = protein_id.split(';')
# 写入文件
for id in ids:
outfile.write(f"{id}\n")
python3 extract.py
sort -n metabolism.pro.id |uniq > rmdup.pro.id #最终得到蛋白ID 4926条
根据蛋白ID从GBFF中提取Gene_id,最终得到基因序列1740条。(测试了不限制物种的注释到pathway,结果为1759条gene)
切割gene id,从ncbi中批量下载gene序列,参考前文,这里不做赘述!
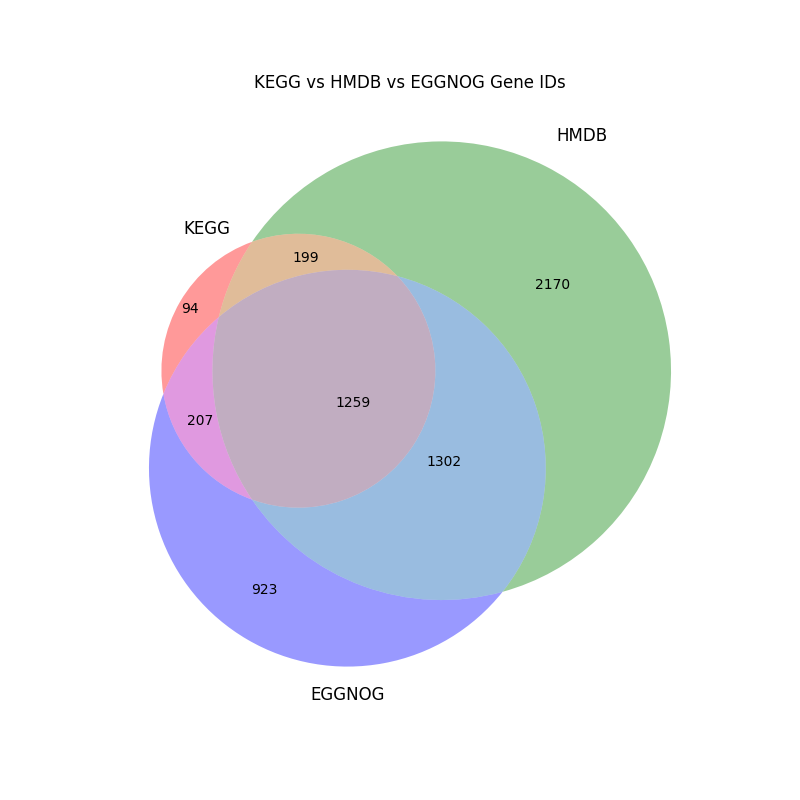
比较三个数据库的注释结果,以韦恩图和表格的形式展示。
先在pc端整理出一个三个数据库注释的gene id表格:all.gene.tab,然后用python分析。
mkdir diff_compare && cd diff_compare
python3 analysis.py
# 读取表格数据
file_path = '/home/xb/1_results/240906_pig_genome_annot/diff_compare/all.gene.tab'
df = pd.read_csv(file_path, sep='\t')
# 将每一列的基因ID提取为集合
kegg_set = set(df['KEGG'].dropna())
hmdb_set = set(df['HMDB'].dropna())
eggnog_set = set(df['EGGNOG'].dropna())
# 生成韦恩图
plt.figure(figsize=(8, 8))
venn = venn3([kegg_set, hmdb_set, eggnog_set], ('KEGG', 'HMDB', 'EGGNOG'))
plt.title('KEGG vs HMDB vs EGGNOG Gene IDs')
output_venn_path = '/home/xb/1_results/240906_pig_genome_annot/diff_compare/venn_diagram.png'
plt.savefig(output_venn_path) # 直接保存图片,不需要显示
plt.close() # 关闭图像窗口
# 提取韦恩图各部分的基因ID
venn_data = {
'KEGG_only': kegg_set - hmdb_set - eggnog_set,
'HMDB_only': hmdb_set - kegg_set - eggnog_set,
'EGGNOG_only': eggnog_set - kegg_set - hmdb_set,
'KEGG_HMDB': kegg_set & hmdb_set - eggnog_set,
'KEGG_EGGNOG': kegg_set & eggnog_set - hmdb_set,
'HMDB_EGGNOG': hmdb_set & eggnog_set - kegg_set,
'All_three': kegg_set & hmdb_set & eggnog_set
}
# 将各部分基因ID保存为表格
output_table_path = '/home/xb/1_results/240906_pig_genome_annot/diff_compare/venn_data.xlsx'
with pd.ExcelWriter(output_table_path) as writer:
for section, genes in venn_data.items():
pd.DataFrame(list(genes), columns=[section]).to_excel(writer, sheet_name=section, index=False)